
More Information
Submitted: 17 July 2019 | Approved: 21 August 2019 | Published: 22 August 2019
How to cite this article: Goodwin D, Olsen AM, Citek K. Unilateral retinitis pigmentosa: Case report and review of the literature. Int J Clin Exp Ophthalmol. 2019; 3: 013-19.
DOI: 10.29328/journal.ijceo.1001021
Copyright Licence: © 2019 de Goodwin D, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Fundus autofluorescence; Optical coherence tomography; Multifocal electroretinography; Retinitis pigmentosa; Unilateral retinitis pigmentosa

Unilateral retinitis pigmentosa: Case report and review of the literature
Denise Goodwin1*, Amanda M Olsen2 and Karl Citek3
1Professor, OD, FAAO, Pacific University College of Optometry, 2043 College Way, Forest Grove, Oregon, USA
2OD, South East Eyecare in Estevan, Saskatchewan, Canada
3Professor, OD, PhD, FAAO, Pacific University College of Optometry, 2043 College Way, Forest Grove, Oregon, USA
*Address for Correspondence: Denise Goodwin, OD, FAAO, Pacific University College of Optometry, 2043 College Way, Forest Grove, OR 97116, USA,Tel: + (503) 352-3070; Fax: (503) 352-2929; Email: [email protected]
Significance: Due to the limited number of reported cases little is known about the characteristics of unilateral retinitis pigmentosa. Information from additional case reports can aid in learning more about the condition. We report a case of retinitis pigmentosa that has remained unilateral for 28 years and review the available literature.
Case Report: A 40-year-old Caucasian female presented for an opinion as to the cause of her vision loss. Fundus autofluorescence demonstrated hypoautofluorescence in the midperipheral retina and a hyperautofluorescent ring surrounding the area of preserved photoreceptors in the macula. Optical coherence tomography showed disruption of the ellipsoid zone and the external limiting membrane. Electroretinography (ERG) showed severely reduced rod and cone function monocularly.
Discussion: Retinitis pigmentosa is typically bilateral and symmetric. Unilateral retinitis pigmentosa is a rare condition that manifests with only one eye having changes typical of retinitis pigmentosa. The unaffected eye can have no signs of retinitis pigmentosa and must have a normal ERG after long-term follow up. It is critical to rule out inflammatory, traumatic, toxic, and cancer associated retinopathy that can present with retinal pigmentary changes. Unilateral retinitis pigmentosa generally remains unilateral, but long-term follow up with ERG is important. There is currently no treatment that can stop the process of retinitis pigmentosa, but gene therapy shows promise.
Retinitis pigmentosa is typically a bilateral, progressive retinal degeneration that ultimately leads to death of both rod and the cone photoreceptors. Retinitis pigmentosa is generally symmetric, but can present asymmetrically [1,2]. A challenge presents if only one eye has the hallmark characteristics of bone spicules in the mid and far periphery of the retina, attenuated retinal vessels, and a pale, waxy appearing optic nerve head. In order to be considered unilateral retinitis pigmentosa, the patient must be followed for a sufficiently long time, at least 5 years, with sensitive testing methods that rule out abnormalities in the contralateral eye [3,4]. Differentiating between bilateral retinitis pigmentosa and the much rarer unilateral retinitis pigmentosa is critical since it affects the prognostic and psychological aspects of management.
There are few reports of unilateral retinitis pigmentosa in the literature, and some have complicating factors or represent extremely asymmetric retinitis pigmentosa [1,5-11]. In retrospective reviews of those with retinitis pigmentosa, between 0.002 and 5% of the study populations had unilateral retinitis pigmentosa [1,3,12]. The higher values12 may be skewed since there was no long term follow up of most of the patients. The true frequency of the disease is unknown, but with more case reports illustrating this disease, additional information may be obtained. Here, we present a case of a woman with unilateral retinitis pigmentosa, and we review the available literature.
A 40-year-old Caucasian female originally presented requesting an opinion as to the cause of her vision loss. After extensive review of past records, we found that she had been given a previous diagnosis of unilateral retinitis pigmentosa by one retinal specialist but was then told she did not have this by another retinal specialist. She started having symptoms at the age of 14 years when she saw a “black, floating oval” in her left eye. Previous exam records indicated that the left eye visual acuity was 20/80 (6/24) twelve years previous to our examination, and 20/200 (6/60) ten years previous to our examination. The vision loss continued to progress to 20/400 (6/120) about 6 years previous to our evaluation. Posterior subcapsular cataract, vascular attenuation, bone spicule pigmentary changes, and cystoid macular edema were noted in the left eye at these visits.
Health history was remarkable for gall bladder surgery and polycystic ovarian syndrome resulting in a hysterectomy. She had no history of trauma or systemic inflammatory conditions but was a former cigarette smoker. She reported no family history of retinitis pigmentosa. She has two children, aged 12 years and 7 years, both of whom had unremarkable ocular health and no evidence of retinitis pigmentosa.
Best corrected visual acuities were 20/15 (6/4.5) in the right eye and light perception at one meter in the left eye. A left constant exotropia of 25 prism diopters was present. Extraocular muscle testing was full with no diplopia or pain on eye movement. Pupils were round and reactive to light with a left relative afferent pupillary defect.
Intraocular pressures were 16 mmHg in each eye. There was grade 2+ posterior subcapsular cataract in the left eye. The remainder of the anterior ocular health was unremarkable in both eyes. Posterior segment in the right eye showed a healthy looking optic nerve head with no pallor and no retinal pigmentary changes. The left eye revealed a pallorous optic nerve head with peripapillary atrophy. The retinal arteries were attenuated. Bone spicules were seen throughout the mid-peripheral retina (Figure 1). A macular lamellar hole was present slightly inferior to the macula.
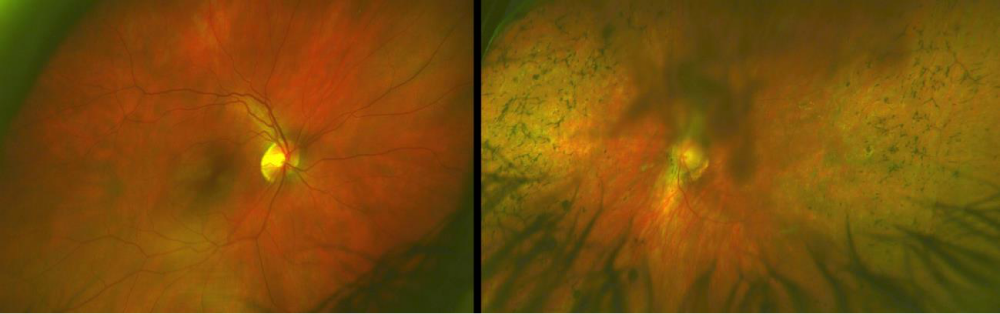
Figure 1: The fundus of the right eye (a) was unremarkable. The left eye image (b) shows bone spicules in the mid-peripheral retina, as well as pallor of the optic nerve and attenuation of the retinal arteries. The left image was partially obstructed due to the posterior subcapsular cataract.
Automated Humphrey 30-2 SITA-standard visual field was unremarkable in the right eye. The left eye showed severe defects in all four quadrants, with relative sparing of the central 5 degrees of field (Figure 2).
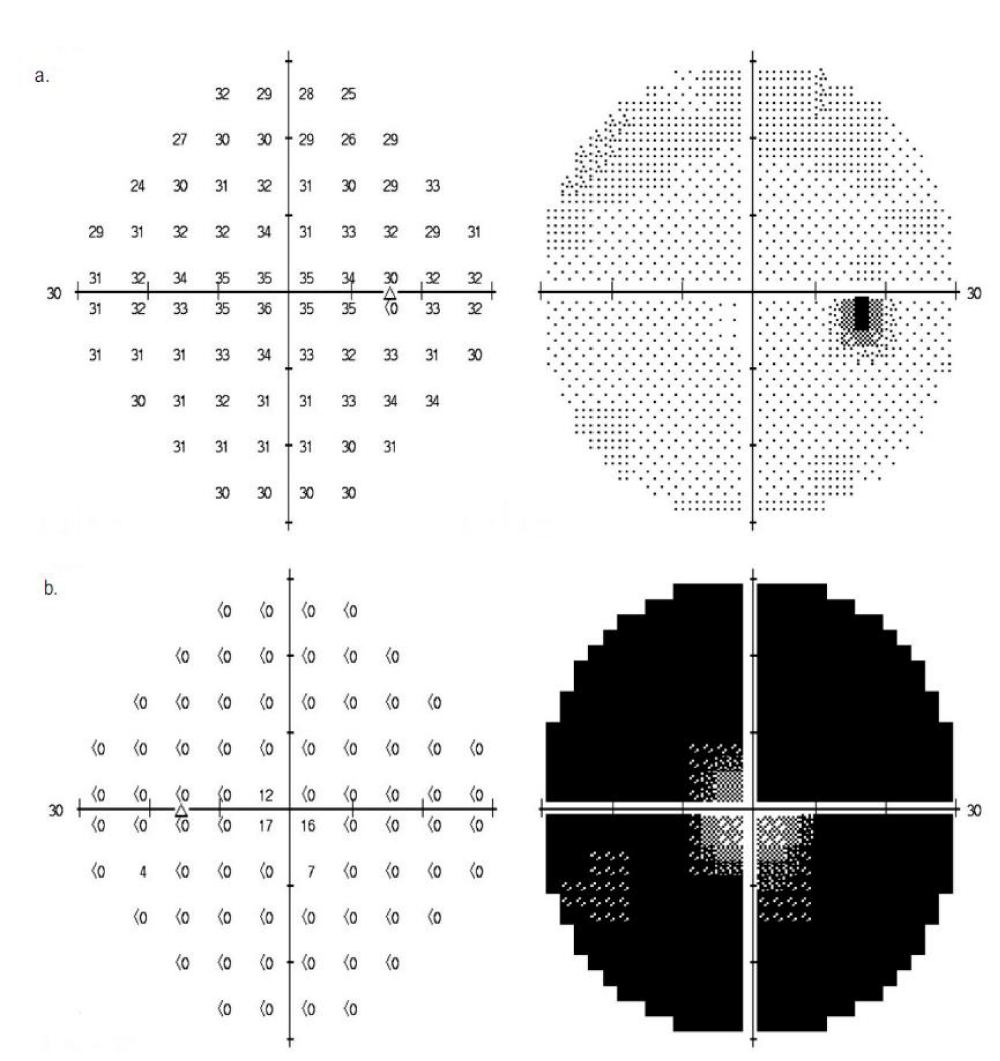
Figure 2: Visual field of the right (a) and left (b) eye.
Macular optical coherence tomography of the right eye was unremarkable with normal macular contour and thickness and no disruption of the photoreceptor inner segment/outer segment junction. The left eye optical coherence tomography revealed a lamellar hole, as well as significant disruption of the inner segment/outer segment junction and external limiting membrane (Figure 3).
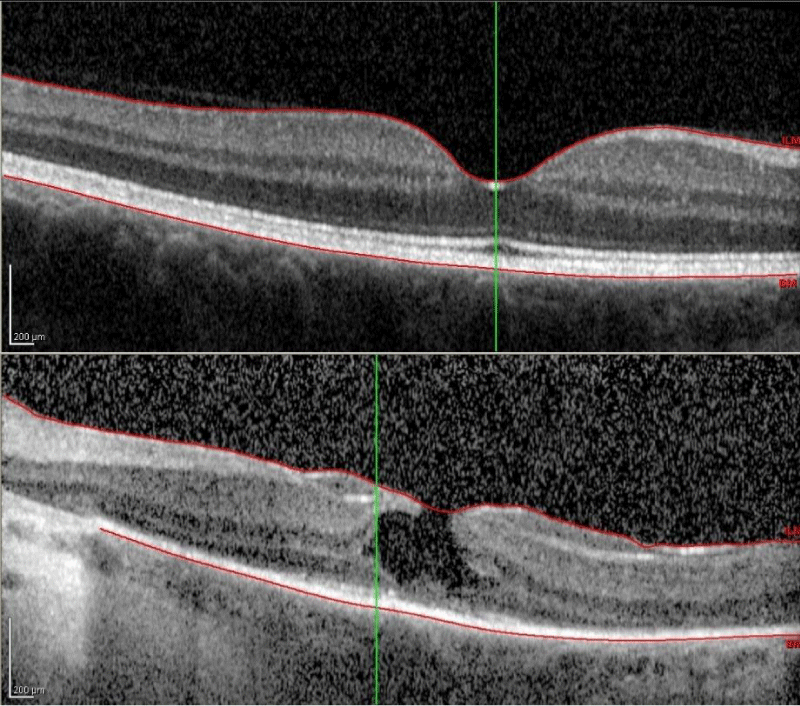
Figure 3: Macular optical coherence tomography of the right eye (top) shows normal foveal architecture and complete photoreceptor inner segment/outer segment junction. Left eye (bottom) demonstrates a partial-thickness lamellar hole, as well as significant disruption of photoreceptor inner segment/outer segment junction, external limiting membrane, and retinal pigmented epithelium.
A 61 Hex multi-focal electroretinography (Diagnosys LLC, Lowell, Massachusetts, USA) performed according to International Society for Clinical Electrophysiology of Vision (ISCEV) standards (http://iscev.org/standards/index.html) with DTL electrodes showed good reliability with normal amplitudes and implicit times in the right eye. The left eye demonstrated decreased amplitudes throughout the central 30 degrees (Figure 4).
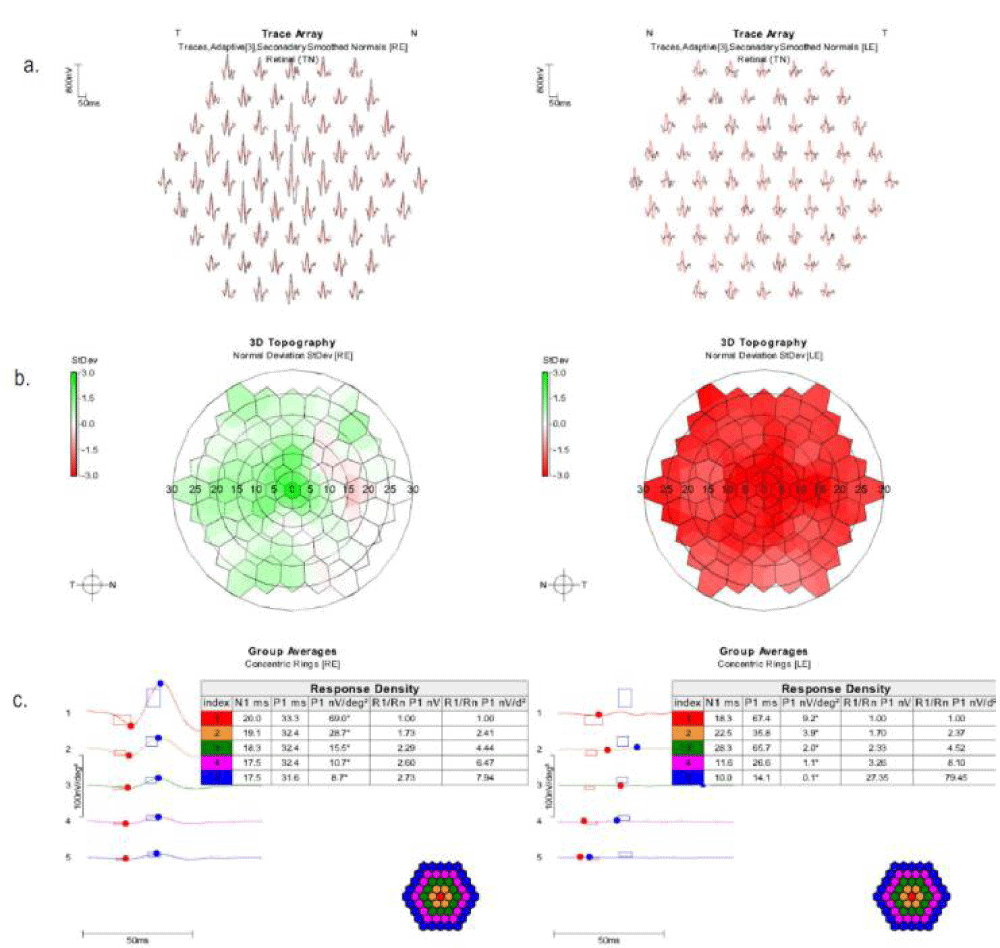
Figure 4: Multifocal electroretinography trace array (a), standard deviation map (b), and group averages (c). Normal trace array values are in red, and patient values are in black. Topography values in red are below average. Those in green are above average.
A full-field electroretinogram (Diagnosys LLC, Lowell, Massachusetts, USA) with DTL electrodes was performed according to ISCEV standards (https://iscev.wildapricot.org/standards). The average of two reliable readings for each protocol is shown in figure 5. Following 10 minutes of light adaptation, the light-adapted 3.0 electroretinogram (30 cd*m-2 background) showed normal a and b wave amplitude and implicit time in the right eye. The left eye demonstrated an indistinguishable curve. This was followed by a light-adapted 3.0 flicker electroretinogram (30 cd*m-2 background) which showed a sinusoidal wave with normal amplitude. The left eye demonstrated an indistinguishable curve. Following 20 minutes of dark adaptation, dark-adapted 0.01, dark-adapted 3.0, and dark-adapted 10.0 electroretinograms were performed. Each showed a well-formed curve with normal a and b wave amplitudes and implicit times for the right eye and an indistinguishable curve in the left eye.
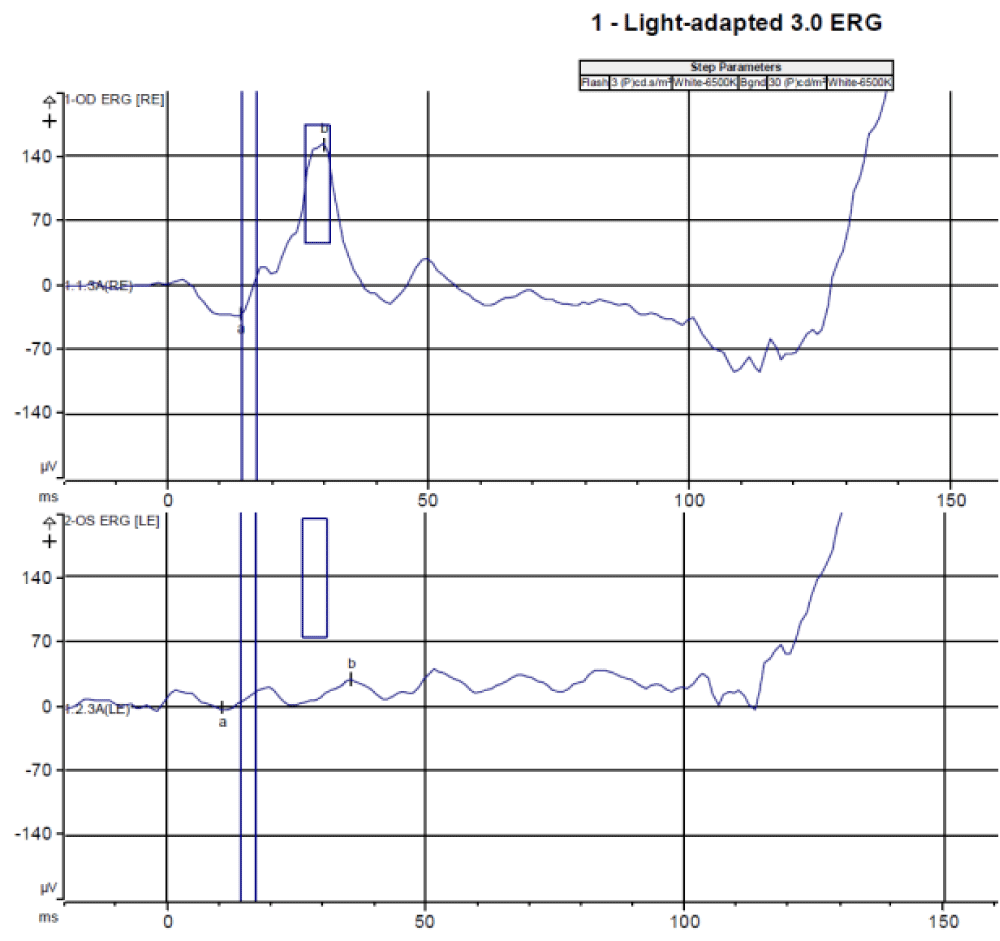
Figure 5: Full field electroretinography showed normal findings in the right eye and significantly reduced function in the left eye with the light-adapted 3.0 electroretinography (a), light-adapted 3.0 flicker electroretinography (b), dark-adapted 0.01 electroretinography (c), dark-adapted 3.0 electroretinography (d), and dark-adapted 10.0 electroretinography.
Fundus autofluorescence showed normal retinal pigmented epithelium in the right eye, but a ring of hyperautofluorescence surrounding the macula, as well as extensive hypoautofluorescence throughout the vascular arcades in the left eye (Figure 6).
We continued to follow the patient for an additional two years with no significant changes. At the most recent visit, her son was tested and did not show any indication of retinitis pigmentosa on electroretinogram.
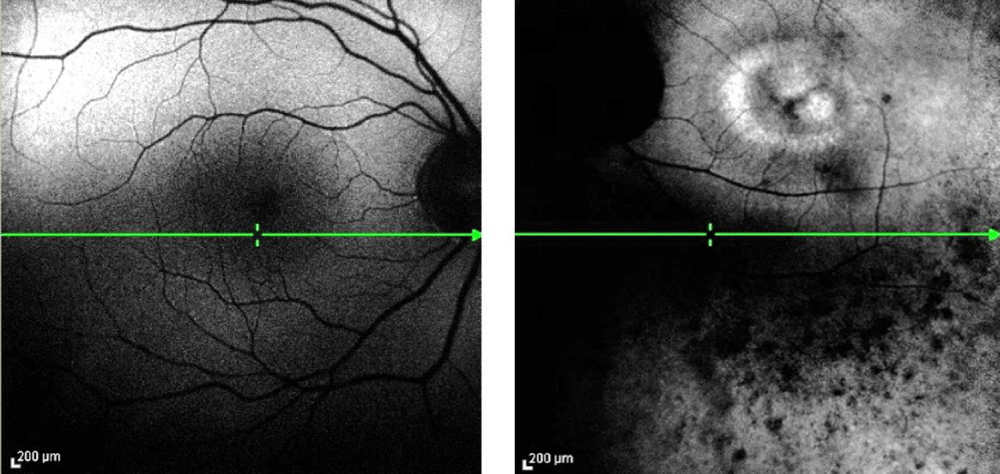
Figure 6: Fundus autofluorescence of the right (a) and left (b) eye. Despite reduced quality due to poor fixation and the cataract, a hyperfluorescent ring is present around the macula, and hypopigmentation is present in the midperipheral retina.
Retinitis pigmentosa is used to describe a group of hereditary retinal disorders that leads to progressive death of retinal photoreceptors, both rods and cones. The classic signs include intraretinal pigmentation (bone spicules) in the mid and far periphery of the retina, attenuated retinal vessels, and a pale, waxy appearing optic nerve head. Other complications associated with retinitis pigmentosa include posterior subcapsular cataracts and cystoid macular edema.
Unilateral retinitis pigmentosa was described as early as 1865 [13]. Since that time the existence of the diagnosis has been questioned. It has been postulated that patients could have had very asymmetric disease that would have been seen if sufficiently sensitive testing was available [14]. Even today the existence of unilateral retinitis pigmentosa is questioned. Some feel bilaterality is an essential diagnostic criteria of retinitis pigmentosa [15]. With the availability of electroretinography, autofluorescence, and optical coherence tomography, confirmation of the diagnosis is now possible.
Epidemiology and etiology
Bilateral retinitis pigmentosa is estimated to affect 1 in 4,000 people and has been widely studied [2,16]. Forty-five separate gene loci have been identified that account for 50% of all bilateral retinitis pigmentosa cases [2]. In addition, between 20-30% of patients with bilateral retinitis pigmentosa have an associated systemic disease, with more than 30 syndromes identified [2]. The most widely known associated syndrome, Usher Syndrome, results in retinitis pigmentosa and hearing impairment.
Unilateral retinitis pigmentosa is reported to occur in 0.002 and 5% of those with retinitis pigmentosa [1,3,12]. To be considered unilateral retinitis pigmentosa, one eye must show functional changes that are typical of retinitis pigmentosa while the other, unaffected eye, can have no symptoms of retinitis pigmentosa and must have a normal electroretinogram [3]. The patient must be examined at sufficiently long intervals to ensure that it is not a highly asymmetric case of bilateral retinitis pigmentosa with delayed onset in the other eye. Inflammatory and other causes of retinal pigmentary changes must also be excluded to satisfy the diagnosis of unilateral retinitis pigmentosa.
The true etiology of unilateral retinitis pigmentosa is difficult to determine due to the small number of cases. Sporadic appearance of the disease is more likely than genetic manifestation in both unilateral and bilateral retinitis pigmentosa. Many case studies suggested that those with unilateral retinitis pigmentosa have no family history of retinitis pigmentosa [1,3,6,11,17]. However, Farrell et al. [12]. Reported that while none of their 14 subjects with unilateral retinitis pigmentosa had a family history of unilateral retinitis pigmentosa, about one third of those with unilateral retinitis pigmentosa had a family member with bilateral retinitis pigmentosa. Unfortunately, there was only long term follow-up on two of these patients so it is possible that these were asymmetric retinitis pigmentosa cases. Regardless, this retrospective study found that of 256 bilateral cases, 34% had a genetic component, with the mode of inheritance being autosomal dominant in 59%, autosomal recessive in 34%, and X-linked in 7%. The 14 unilateral cases had a similar inheritance pattern: 36% of those with unilateral retinitis pigmentosa had a genetic component with 80% being autosomal dominant, 20% being autosomal recessive, and none being X-linked. Mukhopadhyay et al. [18]. presented a case of unilateral retinitis pigmentosa with a p.R677X mutation, the most common cause of autosomal dominant retinitis pigmentosa.
Unilateral retinitis pigmentosa has an age of onset of 31 years for non-familial cases and 38 years for familial cases [12]. This is an older age compared with the age of onset with bilateral retinitis pigmentosa of 11 to 24 years [12,19]. This later age of onset is theorized to be due to the masking of symptoms from the unaffected eye; however, sample sizes in studies of unilateral retinitis pigmentosa are small.
Clinical manifestations/Evaluation
Patients with both bilateral and unilateral retinitis pigmentosa typically present with concerns of decreased night vision or difficulty with dark adaptation. This can be attributed to the death of rod photoreceptors, which in turn causes release of pigment by the retinal pigment epithelial cells creating a bone spicule formation in the mid and far periphery of the retina. The death of these photoreceptors creates a ring-shaped scotoma in peripheral visual field. With time, the rods continue to die, leading to an enlarged scotoma that eventually results in tunnel vision with a small central island of vision. The rod photoreceptors die faster than the cone photoreceptors, which allows relative sparing of the central visual acuity [2]. Other common complaints include decreased peripheral vision and photophobia. Up to 35% experience photopsias [19].
Our patient had all of the classic retinal findings of retinitis pigmentosa, including intraretinal bone spicules, attenuated retinal arterioles, and a pale, waxy optic disc. Other finding consistent with retinitis pigmentosa were present including a posterior subcapsular cataract and evidence of past macular edema (lamellar hole). Other technology helpful in the diagnosis and continuing care of unilateral retinitis pigmentosa includes fundus autofluorescence, optical coherence tomography, and electroretinography.
With fundus autofluorescence, retinal pigmented epithelial cell death is represented by hypoautofluorescence. In retinitis pigmentosa, this hypoautofluorescence is seen throughout midperipheral retina and corresponds with visual field loss [20]. Additionally, more than half of retinitis pigmentosa patients display a hyperautofluorescent ring around the macula [21]. Hyperautofluorescence corresponds to increased levels of lipofuscin in the retinal pigmented epithelium or other fluorophores in the photoreceptor layer [22]. This increased intensity implies stressed retinal pigmented epithelium and ongoing degeneration of photoreceptors. The inner border of the hyperautofluorescent ring has been shown to correspond with the area of preserved visual function and relative preservation of cone photoreceptors on optical coherence tomography [22,23]. Outside the ring, visual function is severely affected [23,24]. The size of the ring may be useful in monitoring the progression of the disease [22,25]. Generally, those with abnormal autofluorescence within the hyperautofluorescent ring have an absent inner segment/outer segment junction in the foveal region [26]. Our patient did have hypoautofluorescence in the midperipheral area, as well as a hyperautofluorescent ring surrounding the macula in the left eye. In addition to the hyperautofluorescent ring, our patient had other areas of hyperautofluorescence within the perimacular ring. This may at least partially account for the severe vision loss.
Optical coherence tomography is useful for evaluating macular edema, but it can also be used in evaluating progression of photoreceptor damage associated with retinitis pigmentosa. The photoreceptor inner segment/outer segment junction is seen on optical coherence tomography as a hyperreflective line above the retinal pigmented epithelium layer. The presence of the inner segment/outer segment junction indicates functioning photoreceptors. Disruption of this line occurs with disturbance of the structure of the cells. In retinitis pigmentosa, the presence and regularity of the inner segment/outer segment junction is correlated with best-corrected visual acuity and preserved visual field, and continued loss of the inner segment/outer segment junction is associated with retinitis pigmentosa progression [27-29]. In addition, the absence the external limiting membrane correlates with decreased visual acuity [27]. This is consistent with our patient where vision was reduced to light perception with absence of the inner segment/outer segment junction and external limiting membrane in the perimacular area and irregular inner segment/outer segment junction in the central retina.
Electroretinography is very helpful in the diagnosis and monitoring of retinitis pigmentosa. Generally, rod function is greatly affected. Cone function, although less affected, is still abnormal. A full field electroretinogram can be extremely helpful in the diagnosis of retinitis pigmentosa as it measures the function of the overall retina. Our patient showed significantly decreased cone and rod function in the left eye. Despite it being more than 25 years since her first symptoms, rod function in the right eye was completely normal.
Multifocal electroretinography may be especially useful in advanced retinitis pigmentosa where the remaining retinal function is too small to be seen with a full field electroretinogram. In mild retinitis pigmentosa, the outer ring average correlates well with the scotopic mixed cone/rod amplitude performed with full field electroretinogram [31]. Multifocal electroretinography shows a gradual loss in amplitude that is greatest in the perimacular area, while more advanced cases also affect the macular response [32]. Implicit time of the central ring remains at a normal level but increases with increased eccentricity [15, 31]. There is a correlation between best-corrected acuity and multifocal electroretinogram amplitude of the central hexagon [31,33]. The relationship between visual acuity and implicit time is more controversial. Moschos et al. [33], found an inverse relationship between acuity and implicit time. Others have not found this relationship between acuity and implicit time [31]. Our patient showed significantly reduced amplitude in all concentric rings, including the central hexagon. Due to the reduced amplitude not forming a definite peak, implicit time is difficult to determine. The central retinal function of this patient may be affected in part due to the presence of the lamellar hole, but given the severity of abnormal central multifocal electroretinogram function, degeneration due to retinitis pigmentosa is more likely to be the cause of our patient’s visual loss.
Differential diagnosis
It is important to rule out other causes of treatable retinal pigmentary degeneration, vessel attenuation, and optic nerve pallor. The main differentials can be excluded with a thorough case history. Infectious or inflammatory conditions, such as syphilis or toxoplasmosis, can result in pigment irregularities similar to retinitis pigmentosa [14,34]. Toxic retinopathies, particularly those caused by phenothiazine drugs, chloroquine, or hydroxychloroquine can cause maculopathy, peripheral pigmentary retinopathy, and decreased night vision [6]. Traumatic retinopathies, especially those from blunt trauma, can also show retinal pigmentary changes similar to retinitis pigmentosa. Finally, cancer associated retinopathy can present with similar signs such as retinal artery attenuation and pigmentary changes. However, the time course for vision loss occurs over week or months for cancer associated retinopathy compared to years with retinitis pigmentosa. Our patient reported no history consistent with inflammation, use of known retinotoxic medications, or trauma; and the vision loss occurred over many years.
Treatment/Management
There are currently no studies involving the treatment of unilateral retinitis pigmentosa so management is based on bilateral retinitis pigmentosa studies. Unfortunately, there is currently no treatment that can stop or reverse the process of retinitis pigmentosa. Gene therapy is being investigated as a treatment, and in studies with Leber congenital amaurosis it has been shown to be protective rather than restorative [30]. Because of this, gene therapy is likely more effective in the early stages of the disease. Vitamin A therapy is controversial, and the minimal slowing of retinitis pigmentosa progression must be balanced by the potential adverse effects of long-term, high dosage vitamin A usage [30,31]. Other antioxidants, including omega-3 fatty acid and lutein, may have some benefit, but more studies are necessary to determine the effectiveness. With bilateral retinitis pigmentosa, low vision services are often utilized, but this may not be necessary in unilateral retinitis pigmentosa depending on the quality of vision in the unaffected eye. Genetic counseling is recommended, especially if there is a family history of retinitis pigmentosa. Cataract extraction may be warranted to decrease glare and improve vision of the affected eye, although prognosis is guarded in those with a disruption of the inner segment/outer segment junction [32].
Prognosis
There are very few studies on long-term follow-up of unilateral retinitis pigmentosa, but it has been previously understood that unilateral retinitis pigmentosa stays unilateral. Farrell et al. [12], had two patients who were followed for 8 years and 14 years, respectively, and in both cases the unaffected eye had no signs of RP. Weller et al. [5], had one patient who showed no changes in the unaffected eye over a time span of 30 years. However, Gauvin and colleagues [33], had a patient present with unilateral retinitis pigmentosa who was seen eight separate times over thirty years during which bilateral retinitis pigmentosa developed. The electroretinogram was the first assessment to detect the development of signs of retinitis pigmentosa in the contralateral eye. There should be caution in explaining long-term prognosis to patients with unilateral retinitis pigmentosa. Follow-up visits, especially electroretinography, should be conducted routinely with these patients.
Most of what is known about the progression of unilateral retinitis pigmentosa is inferred from studies on bilateral retinitis pigmentosa. However, Potsidis et al. [3], retrospectively investigated 15 cases of unilateral retinitis pigmentosa to determine the rate of visual field and electroretinogram loss compared to the normal, age-related changes of the opposite eye. The visual field had an annual decrease of 4.9% compared to no change in a normal, aging eye. The electroretinogram amplitude with 0.5 Hz flashes decreased by 4.7% each year, whereas senile changes resulted in a decrease of 1.4%. The electroretinogram amplitude with 30.0 Hz flashes showed a similar decrease of 4.6% each year in the retinitis pigmentosa eye versus 1.2% decrease in the normal, aging eye.
Here we present a case of unilateral retinitis pigmentosa. Twenty-eight years following the initial symptoms, a complete workup showed no signs of retinitis pigmentosa in the contralateral eye. Unilateral retinitis pigmentosa is a rare condition in which only one eye manifests the symptoms and clinical hallmarks of retinitis pigmentosa. The unaffected eye has no signs or symptoms of retinitis pigmentosa, including a normal electroretinogram. Once the differential diagnoses are ruled out, electrodiagnostic data can aid in confirming the diagnosis of unilateral retinitis pigmentosa. More information about the progression of this disease is required and patients with unilateral retinitis pigmentosa should be closely monitored for changes in the unaffected eye. Immediate family members, especially children, should be assessed as well in order to establish or rule out inheritance patterns.
- Auerbach E, Rowe H. The “good” eye in unilateral retinitis pigmentosa. Ophthalmologica. 1968; 155: 98-116. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/5636434
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006; 368: 1795-1809. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17113430
- Potsidis E, Berson EL, Sandberg MA. Disease course of patients with unilateral pigmentary retinopathy. Invest Opthalmology Vis Sci. 2011; 52: 9244-9249. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21989720
- Francois J, Verriest G. Retinitis pigmentosa unilateral. Ophthalmologica. 1952; 124: 65-88. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12993447
- Weller JM, Michelson G, Juenemann AG. Unilateral retinitis pigmentosa: 30 years follow-up. BMJ Case Rep. 2014; bcr2013202236-bcr2013202236. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24515232
- Brill TF. Marcus Gunn pupil in a possible case of unilateral retinitis pigmentosa. Am J Optom Physiol Opt. 1979; 56: 252-258. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/517626
- Marsiglia M, Duncker T, Peiretti E, Brodie SE, Tsang SH. Unilateral retinitis pigmentosa: a proposal of genetic pathogenic mechanisms. Eur J Ophthalmol. 2012; 22: 654-660. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22139616
- Pearlman JT, Saxton J, Hoffman G. Unilateral retinitis pigmentosa sine pigmento. Br J Ophthalmol. 1976; 60: 354-360. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1042728/
- Alina-Cristina S, Marian B, Mihail Z. Unilateral pigmentary retinopathy – a review of literature and case presentation. Rom J Ophthalmol. 2016; 60: 47-52. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27220234
- Thakur A, Puri L. Unilateral retinitis pigmentosa. Clin Exp Optom. 2010; 93: 102-104. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20406260
- Kolb H, Galloway NR. Three cases of unilateral pigmentary degeneration. Br J Ophthalmol. 1964; 48: 471-479. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14218207
- Farrell DF. Unilateral retinitis pigmentosa and cone-rod dystrophy. Clin Ophthalmol. 2009; 3: 263-270. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19668577
- Padraglia C. Klinische beobachtungen. Retinitis pigmentosa. Klin Mbl Augenheilk. 1865; 3: 114-117. PubMed: https://www.ncbi.nlm.nih.gov/ pubmed/3259404
- Skalka HW. Asymmetric retinitis pigmentosa, luetic retinopathy and the question of unilateral retinitis pigmentosa. Acta Ophthalmol (Copenh). 1979; 57: 351-357. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/474081
- Seeliger M, Kretschmann U, Apfelstedt-Sylla E, Rüther K, Zrenner E. Multifocal electroretinography in retinitis pigmentosa. Am J Ophthalmol. 1998; 125: 214-226. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9467449
- Bunker CH, Berson EL, Bromley WC, Hayes RP, Roderick TH. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol. 1984; 97: 357-365. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/6702974
- Spadea L, Magni R, Rinaldi G, Dragani T, Bianco G. Unilateral retinitis pigmentosa: clinical and electrophysiological report of four cases. Ophthalmologica. 1998; 212: 350-354. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9693297
- Mukhopadhyay R. Holder GE, Moore AT, Webster AR. Unilateral retinitis pigmentosa occurring in an individual with a germline mutation in the rp1 gene. Arch Ophthalmol. 2011; 129: 954-956. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21746989
- Heckenlively JR, Yoser SL, Friedman LH, Oversier JJ. Clinical findings and common symptoms in retinitis pigmentosa. Am J Ophthalmol. 1988; 105: 504-511. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3259404
- Ogura S, Yasukawa T, Kato A, Usui H, Hirano Y, et al. Wide-field fundus autofluorescence imaging to evaluate retinal function in patients with retinitis pigmentosa. Am J Ophthalmol. 2014; 158: 1093-1098. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25062603
- Mitamura Y, Mitamura-Aizawa S, Nagasawa T, Katome T, Eguchi H, et al. Diagnostic imaging in patients with retinitis pigmentosa. J Med Invest. 2012; 59: 1-11. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22449988
- Greenstein VC, Duncker T, Holopigian K, Carr RE, Greenberg JP, et al. Structural and functional changes associated with normal and abnormal fundus autofluorescence in patients with retinitis pigmentosa. Retina. 2012; 32: 349-357. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21909055
- Escher P, Tran HV, Vaclavik V, et al. Double concentric autofluorescence ring in NR2E3-p.G56R-linked autosomal dominant retinitis pigmentosa. Investig Opthalmology Vis Sci. 2012; 53: 4754-4764. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22661467
- Duncker T, Tabacaru MR, Lee W, Tsang SH, Sparrow JR. Comparison of near-infrared and short-wavelength autofluorescence in retinitis pigmentosa. Investig Opthalmology Vis Sci. 2013; 54: 585-591. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23287793
- Lima LH, Burke T, Greenstein VC, Chai LC, Wener C, et al. Progressive constriction of the hyperautofluorescent ring in retinitis pigmentosa. Am J Ophthalmol. 2012; 153: 718-727. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3830946/
- Iriyama A, Yanagi Y. Fundus autofluorescence and retinal structure as determined by spectral domain optical coherence tomography, and retinal runction in retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2012; 250: 333-339. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21947266
- Battaglia Parodi M, La Spina C, Triolo G, Riccieri F, Pierro L, et al. Correlation of SD-OCT findings and visual function in patients with retinitis pigmentosa. Graefes Arch Clin Exp Ophthalmol. 2016; 254: 1275-1279. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26472300
- Hagiwara A, Mitamura Y, Kumagai K, et al. Photoreceptor impairment on optical coherence tomographic images in patients with retinitis pigmentosa. Br J Ophthalmol. 2013; 97: 237-238. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23172877
- Yoon CK, Yu HG. The structure-function relationship between macular morphology and visual function analyzed by optical coherence tomography in retinitis pigmentosa. J Ophthalmol. 2013; 2013: 1-7. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3866715/
- Guadagni V, Novelli E, Piano I, Gargini C, Strettoi E. Pharmacological approaches to retinitis pigmentosa: a laboratory perspective. Prog Retin Eye Res. 2015; 48: 62-81. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26113212
- Rayapudi S, Schwartz SG, Wang X, Chavis P. Vitamin A and fish oils for retinitis pigmentosa. Cochrane Database Syst Rev. 2013; 12: CD008428. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24357340
- Yoshida N, Ikeda Y, Murakami Y, Nakatake S, Fujiwara K. Factors affecting visual acuity after cataract surgery in patients with retinitis pigmentosa. Ophthalmology. 2015; 122: 903-908. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25601536
- Gauvin M, Chakor H, Koenekoop RK, Little JM, Lina JM, et al. Witnessing the first sign of retinitis pigmentosa onset in the allegedly normal eye of a case of unilateral rp: a 30-year follow-up. Doc Ophthalmol. 2016; 132: 213-229. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27041556